510(k) Submission Platform
Professional FDA 510k services and 510(k) submission services powered by a chain of specialized AI agents. Complete platform from device classification to FDA clearance. Maximize quality, eliminate errors and oversights, while dramatically reducing time and effort required for your submission.
4-5 hours. Complete workflow. Expert guidance.
Funding launch promo: 90% off for 3 months (through May 21, 2026).
What teams say
Real outcomes from teams using Cruxi for 510(k) work.
Cruxi delivered a highly insightful report that proved valuable for our work, even as a few initial technical issues were being resolved.
The 510(k) workflow saved us thousands of dollars and time and significantly enhanced the quality of submission.
The Cruxi Way: Maximum Quality, Zero Risk
Start with Cruxi's AI agents. Finish with expert review. Get two consultants—AI and human—for maximum quality assurance.
Cruxi AI Drafts Your Complete 510(k)
4-5 hours
- check_circleEnter your device information and follow a guided workflow
- check_circleTrained specialized agents identify device classification, regulation number, and product code
- check_circleTrained predicate analysis agent evaluates and scores potential predicate devices
- check_circleTrained regulatory assessment agent maps requirements across all 18 eSTAR sections
- check_circleTrained reasoning agent drafts submission sections with traceable source citations
- check_circleComplete evidence checklist and submission package generation
Expert Review & Finalization
Optional but Recommended
- check_circleVetted FDA consultant reviews your complete Cruxi draft
- check_circleExpert identifies gaps, corrects issues, and improves FDA submission readiness
- check_circleFinal quality assurance to reduce RTA (Refuse to Accept) risk
- check_circleConsultant reviews and finalizes the package for FDA submission readiness
Saves 30+ hours of consultant time. Expert starts from a complete, structured draft instead of a blank page.

Start with a free device assessment (no sign up required)
Your device information and project materials are kept confidential and stay within Cruxi's platform and AI workflow unless you explicitly authorize sharing with a provider for a quote or human review. Privacy Policy
We'll connect you to a vetted consultant once your Cruxi project is ready. Any human consultant review or follow-on work is an independent professional service provided directly by the consultant, not by Cruxi. Cruxi facilitates communication and maintains a directory of qualified experts, and you're also free to work with any consultant you know outside the Cruxi network.
Why This Approach?
Two Experts arrow_downward Time
You get both AI and human expertise. Our specialized AI agents act as your first consultant, doing the heavy lifting. Then a human expert reviews and finalizes—ensuring nothing is missed.
Up to 80% Cost Savings arrow_downward Cost
Since consultants only review an already-drafted submission (not build from scratch), their time is focused on quality assurance. This dramatically reduces costs while maximizing quality.
Zero-Risk Quality Assurance arrow_upward Quality
Expert review catches gaps, prevents RTA issues, and ensures FDA acceptance. The consultant's role is limited to review and refinement—leaving no room for mistakes.
Need a human expert? Cruxi can get you quotes from vetted experts in mins.
Explore 100+ vetted 510k submission expertsassessment Not Sure Where to Start? Get a Free 510(k) Device Assessment
Get instant regulatory insights for your medical device. Enter your device information and receive a quick assessment including classification, regulatory pathway, timeline estimates, and key requirements—all free, no signup required.
Try Free Assessment NowNo signup required • Instant results • No credit card needed • 1 min to complete
Need a detailed 510(k) regulatory pathway assessment?
If you need a comprehensive written regulatory strategy, product code options, predicate landscape, and testing roadmap before committing to a full 510(k) project, our 1-day 510(k) Regulatory Pathway Assessment & Roadmap provides detailed analysis and documentation you can share with your team or consultant.
Learn About the 510(k) Pathway Assessment ($300)Complete 510(k) Submission Workflow
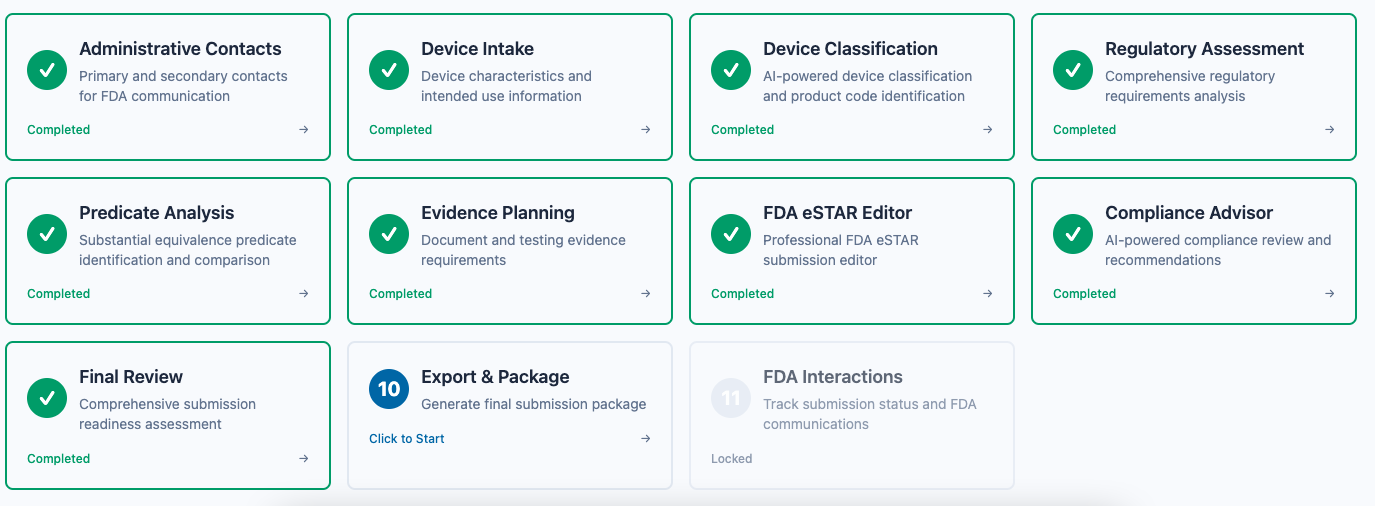
From initial device intake to FDA clearance—every step automated and validated.
Step-by-Step Submission Process
Our guided workflow takes you through each phase of your 510(k) submission, ensuring nothing is missed. From administrative contacts to final export, every step is validated and optimized for FDA acceptance.
- personAdministrative Contacts & Device Intake
- tagAI-Powered Device Classification
- assessmentComprehensive Regulatory Assessment
- searchPredicate Device Analysis
- folder_managedEvidence Planning & Upload
- descriptioneSTAR Section Drafting & Review
- check_circleFinal QA & Submission Package
510(k) Microservices
Powerful standalone tools that work together seamlessly. Use individual microservices or the complete workflow.
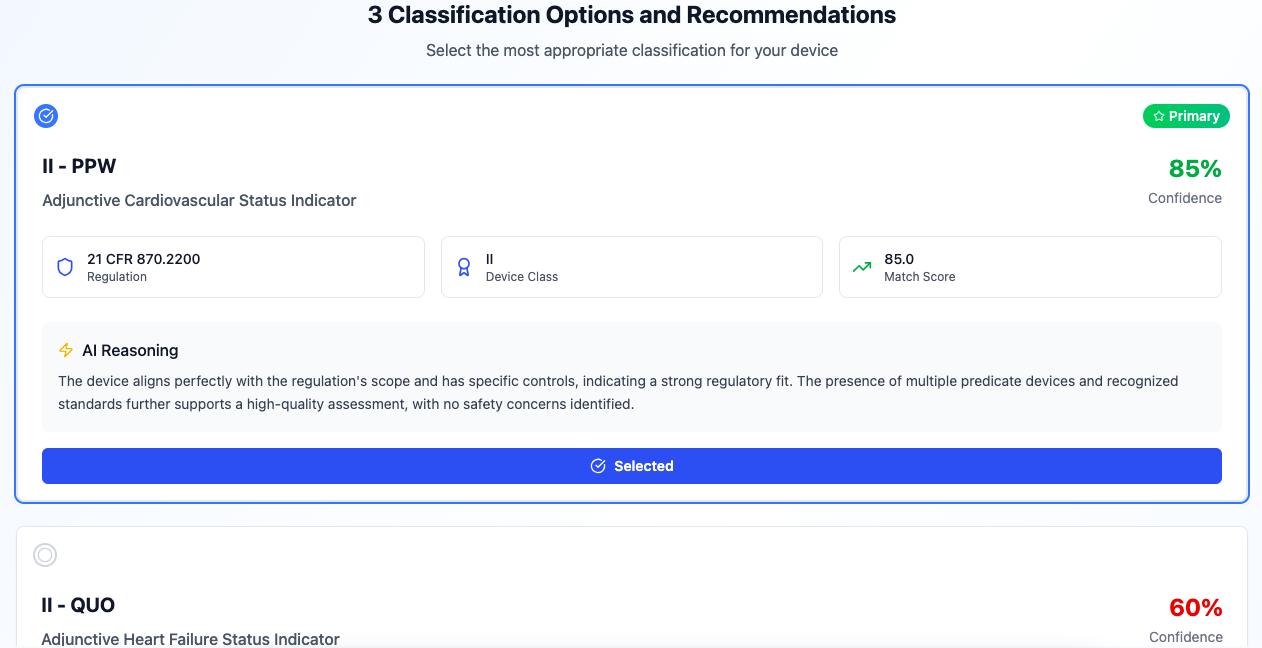
Device Classification
AI-powered device classification with comprehensive analytics. Instantly identify product codes, regulation numbers, and classification pathways. Compare against thousands of cleared devices to find the right classification for your medical device.
- auto_awesomeAutomated product code identification
- searchCompare against cleared device database
- analyticsComprehensive classification analytics
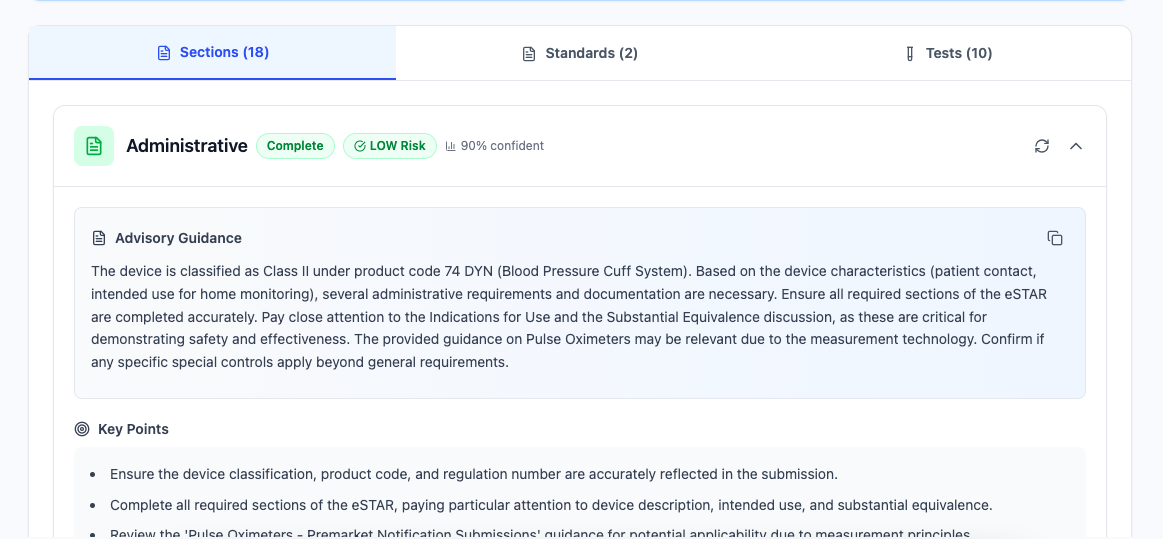
Regulatory Assessment
Comprehensive regulatory requirements analysis across all 18 eSTAR sections. Our AI-powered sectionator analyzes your device and generates intelligent content for each required section, ensuring compliance with FDA guidance.
- assessment18 eSTAR sections analyzed
- smart_toyAI-powered content generation
- verifiedFDA guidance compliance
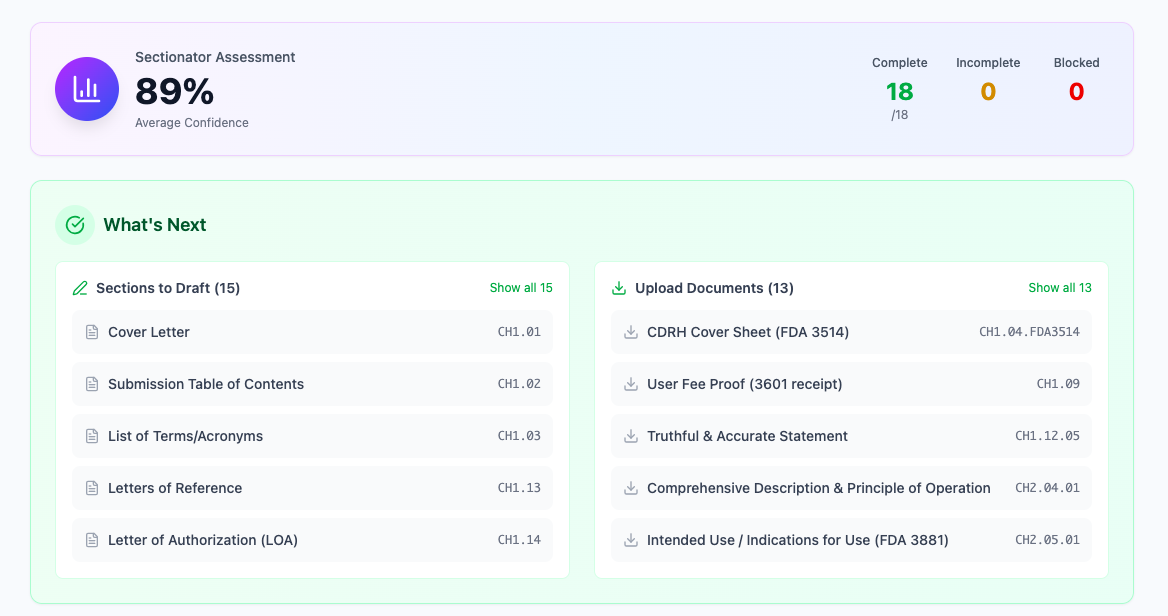
Detailed Section Analysis
Deep dive into each eSTAR section with intelligent content generation. Our system understands FDA reviewer expectations and structures content to facilitate review, reducing the likelihood of additional information requests.
- psychologyFDA reviewer psychology insights
- descriptionSection-by-section breakdown
- checklistCompliance validation
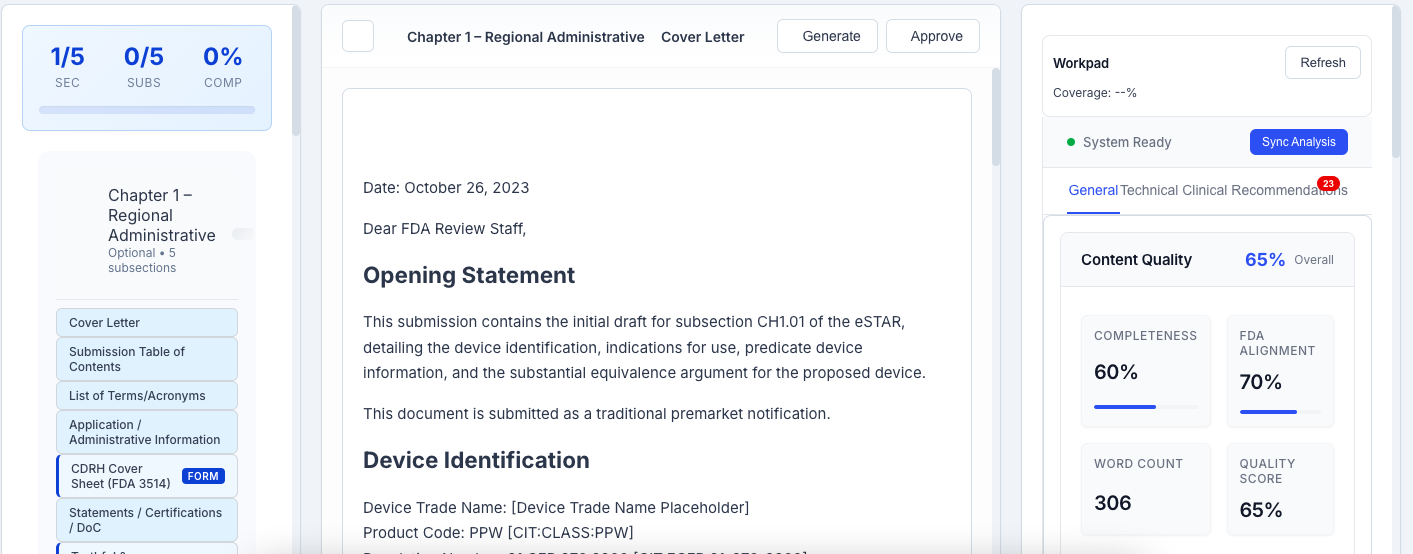
eSTAR Editor
Professional FDA eSTAR submission editor with intelligent drafting, review, and export capabilities. Edit sections with AI assistance, track changes, and generate submission-ready packages formatted for FDA eSTAR submission.
- editIntelligent drafting assistance
- historyTrack changes and revisions
- downloadExport submission-ready packages
Submission Package
Generate complete submission packages with all required documents, properly formatted and organized. Our system ensures all attachments, forms, and supporting documents are correctly structured for FDA submission.
- folder_managedComplete document organization
- ruleFDA format compliance
- check_circleReady for submission
Why Choose Cruxi for Your 510(k)
Our platform combines AI intelligence with regulatory expertise to deliver submissions that meet the highest quality standards.
Cruxi isn't traditional compliance software. It's a regulatory engine that analyzes FDA data, maps your device, and generates FDA-ready 510(k) content — not just a place to store documents and tick checkboxes.
Automated RTA Prevention
Catch issues before submission
Zero Hallucinations
Every claim traceable to source
FDA Reviewer Psychology
Content structured for easy review
Real-Time Updates
Always current with latest FDA guidance
Expert Review
Optional human validation at critical points
Continuous Improvement
Learns from every submission
How Cruxi Works: AI Agents & Comprehensive FDA Database
Behind every 510(k) submission, specialized AI agents do the heavy lifting—from classification to assessment to predicate analysis—while you and your regulatory experts stay in full control. All of this is powered by our up-to-date, well-classified FDA regulatory database so decisions are grounded in real FDA data, not generic AI guesses.
Specialized AI Agents Handle the Heavy Lifting
Unlike generic AI tools, Cruxi uses purpose-built agents that understand FDA regulations, device classification, and submission requirements. Each agent is trained on regulatory expertise and FDA guidance.
auto_awesome NucAgent: Device Classification
Our trained classification agent analyzes your device description, intended use, and technology to identify the correct product code, regulation number, and device class. It searches through thousands of cleared devices, regulations, and FDA guidance documents to provide comprehensive classification analytics with confidence scores.
search PreAgent: Predicate Analysis
Our trained predicate analysis agent evaluates potential predicate devices for substantial equivalence. It scores predicates across multiple dimensions—intended use similarity, technological characteristics, performance data, and regulatory history—providing FDA acceptance probability, cost breakdowns, and time-to-market impact analysis.
psychology Reasoning Agent: Regulatory Assessment & Drafting
Our trained reasoning agent analyzes all 18 eSTAR sections, generates intelligent content, and drafts submission sections. It understands FDA reviewer psychology, structures content for easy review, and ensures every claim is traceable to source documents—preventing hallucinations and ensuring compliance.
shield_person Built for High-Stakes Devices, Not AI Experiments
Cruxi is designed for teams investing millions in development and market access. The AI never "decides" your regulatory strategy or files anything with FDA. It creates structured, evidence-linked drafts that always require human review and sign-off from you and your consultants.
- Human-in-the-loop by design: Every classification, predicate suggestion, and draft section is an assistive recommendation, not an automated decision.
- Traceable to sources: All key statements are grounded in either FDA regulations/guidance, public 510(k) summaries, or your own uploaded documents—never invented citations.
- No black box surprises: You can see why a recommendation was made (e.g., matching indications, product codes, or specific guidance) and accept, modify, or reject it.
- Your data stays yours: Device information and documents are stored securely and used to serve your project—not to train a public model.
Comprehensive, Up-to-Date FDA Regulatory Database
All our AI agents are powered by a comprehensive FDA regulatory database that we maintain, classify, and update regularly. This isn't just raw data—it's well-structured, cross-referenced, and enriched with metadata for intelligent search and analysis.
Product Codes
Complete catalog of FDA product codes with definitions, device classes, exemption status, and regulation mappings
Regulations
Full text of 21 CFR regulations with cross-references to product codes, guidances, and standards
510(k) Clearances
Database of cleared devices with summaries, predicates, product codes, and clearance dates
FDA Guidance Documents
Comprehensive collection of FDA guidance documents, special controls, and regulatory requirements
Recognized Standards
IEC, ISO, and other recognized standards mapped to product codes and regulations
Safety Data
Recalls, MAUDE events, and adverse event data linked to product codes for risk analysis
Regular Updates
Database is continuously updated with new clearances, guidances, and regulatory changes
Predicate Devices
Tens of thousands of cleared 510(k) devices with full summaries, predicates, and substantial equivalence data for comprehensive matching
Special Controls
Comprehensive mapping of special controls, classification panels, and exemption statuses linked to product codes and regulations
Why This Matters: This keeps Cruxi's output grounded in what actually exists in FDA records and your own files—not in hallucinated text or generic internet training data.
How Cruxi Radically Enhances Quality
Discover the 10 superhuman capabilities that set Cruxi apart from traditional consulting and manual processes
What Cruxi Does
Scans ~2,300+ regulations and thousands of guidance/special controls in seconds to see what might apply to your device.
Why No Human Can Match This
A human has to search, open, read, and interpret each source manually. Doing this exhaustively is practically impossible under time and budget pressure.
Quality & Business Impact
You don't "miss a regulation" because someone forgot a keyword. Stronger compliance baseline, fewer blind spots.
What Cruxi Does
Matches your device against thousands of product codes and 10,000s of cleared devices with complex algorithms (indications, tech, risk) to shortlist the best predicates.
Why No Human Can Match This
Humans scan a handful of devices and rely on experience and Google/510(k) search. Nobody can manually compare against tens of thousands consistently.
Quality & Business Impact
Much better predicate selection: tighter similarity, less FDA pushback, fewer AI/RTA questions, lower NSE risk.
What Cruxi Does
Evaluates MAUDE events, recalls, safety communications and recent clearances together when ranking predicates and shaping your strategy.
Why No Human Can Match This
Humans rarely have time to deeply cross-check adverse events and recalls for every candidate predicate, let alone in real time.
Quality & Business Impact
Avoid "toxic" predicates with bad post-market history. Better risk story, safer device positioning, and fewer surprises later.
What Cruxi Does
Generates a device-specific testing matrix in seconds (bench, software, EMC, biocomp, electrical safety, usability, cybersecurity, etc.) mapped to regulations, guidance, and standards.
Why No Human Can Match This
Manually stitching this together from regulations, guidance, and dozens of standards is slow, error-prone, and often inconsistent across projects.
Quality & Business Impact
More complete and defensible evidence plan; reduced risk of FDA asking "Where is the testing for X?" in an AI request.
What Cruxi Does
Runs full-document consistency checks across your submission (IFU, labels, Form 3881, comparison tables, risk files, test reports) to catch wording drift and contradictions.
Why No Human Can Match This
Humans skim; even great reviewers miss small wording differences across 200+ pages, especially under deadline.
Quality & Business Impact
Fewer inconsistencies for FDA to latch onto. Polished, "tells one story" submission quality.
What Cruxi Does
Maintains a live, cross-linked knowledge graph of your device: indications → risks → requirements → tests → results → labeling.
Why No Human Can Match This
A person might keep a spreadsheet or diagram, but manually updating every link with each change is not realistic.
Quality & Business Impact
Strong traceability story; easier answers to FDA questions like "Where do you show that risk X is mitigated and verified?"
What Cruxi Does
Simulates "what if" scenarios instantly – e.g., changing indication wording, removing a test, adding a feature – and shows regulatory/testing impact.
Why No Human Can Match This
Humans need hours or days to re-assess impact, read new guidance, and update documents each time.
Quality & Business Impact
Faster iteration, better decisions, less "accidental" scope creep into De Novo/PMA territory.
What Cruxi Does
Continuously learns from every project run through Cruxi (patterns of FDA questions, AI/RTA issues, testing expectations) and feeds that back into future recommendations.
Why No Human Can Match This
Individual consultants learn from their own cases; teams rarely aggregate and analyze hundreds of submissions' patterns at scale.
Quality & Business Impact
Institutionalized "collective experience" that keeps getting better instead of walking out the door with individuals.
What Cruxi Does
Scores draft sections against FDA expectations (clarity, completeness, alignment with guidance and predicates) before humans ever touch them.
Why No Human Can Match This
A human reviewer can give qualitative feedback, but cannot instantly benchmark every paragraph against thousands of prior examples.
Quality & Business Impact
Higher first-pass quality, fewer review cycles, more time for humans to focus on judgment, strategy and negotiation.
What Cruxi Does
Works 24/7 at full speed: scanning, checking, re-running analyses every time you tweak the device profile or add new data.
Why No Human Can Match This
Humans get tired, context-switch, go on vacation, and can only hold so much in working memory.
Quality & Business Impact
Stable, always-on quality baseline; reduced dependence on single "hero" reviewers.
The Punchline
Cruxi does what no human team can realistically do at scale or speed: mass-scanning regulations, codes, predicates, recalls, MAUDE events, and guidance in seconds and without getting tired.
But Cruxi is not a replacement for experts.
Cruxi + experienced human regulatory professionals = the highest quality 510(k) a company can produce.
You bring judgment, ethics, and real-world context. Cruxi brings exhaustive coverage, speed, and consistency. Together, you get safer devices, stronger submissions, and fewer expensive surprises.
Frequently Asked Questions
Common questions about FDA 510(k) traditional submissions. Cruxi is built for teams who don't have room for AI experiments on a multimillion-dollar device program.
What is a 510(k) traditional submission?
A 510(k) traditional submission is the standard pathway for medical device clearance when demonstrating substantial equivalence to a legally marketed predicate device. It requires comprehensive documentation including device description, intended use, technological characteristics, and clinical data.
How long does a 510(k) submission take?
FDA's standard review time for 510(k) submissions is 90 calendar days. However, preparation time varies significantly. With Cruxi, you can find classification, predicate, and assessment in 5 hours. Once your evidence is complete, you can draft and package eSTAR in 2 hours.
What's the difference between traditional and eSTAR submission?
Traditional 510(k) submissions use PDF documents, while eSTAR (electronic Submission Template And Resource) is FDA's structured electronic format. eSTAR submissions are now required for most device types and offer faster review times. Our platform supports both formats and can convert traditional submissions to eSTAR.
How does AI help with 510(k) submissions?
Cruxi is a complex system of trained multi-agent AI designed to do regulatory work based on up-to-date FDA data, exceeding human quality in many aspects. Our specialized agents automate device classification, identify predicate devices, generate regulatory assessments, and draft eSTAR sections. Nonetheless, human review and final touches are recommended, especially for teams not experienced with FDA submissions. This ensures the highest quality and compliance with current FDA guidance while maintaining full traceability to source documents.
What documents are required for a 510(k) submission?
Required documents include device description, intended use statement, indications for use, device labeling, substantial equivalence comparison, performance testing data, biocompatibility data (if applicable), software documentation (if applicable), and clinical data (if required). Our platform guides you through each requirement.
How much does a 510(k) submission cost?
FDA user fees for 510(k) submissions are approximately $21,000 (as of 2025). Additional costs include testing, clinical studies (if needed), and regulatory consulting. Our platform reduces costs by up to 80% by leveraging the power of specialized AI agents while maximizing quality significantly.
What 510k services does Cruxi provide?
Cruxi provides end-to-end 510k services, including device classification, predicate analysis, regulatory assessment across all eSTAR sections, evidence planning, AI-drafted eSTAR content, and submission package generation. You can use the full 510k submission service or individual microservices (classification, predicate, assessment, eSTAR drafting) depending on your needs.
How does the Cruxi workflow process work?
Cruxi guides you through a step-by-step workflow: First, you enter your device information and administrative contacts. Our AI then classifies your device and suggests predicate devices. Next, the platform generates a comprehensive regulatory assessment across all 18 eSTAR sections, mapping required evidence and drafting content. You upload your evidence documents, and Cruxi structures everything into a submission-ready package. Throughout the process, you can add a consultant for review and finalization, or export the package to submit yourself.
How do I get started with Cruxi?
Getting started is simple: Sign in with your Google account to create a free project. Choose your path—use Cruxi independently, start with Cruxi and add expert review later, or speak with a consultant first. Once you begin, you'll be guided through each step with clear instructions. You can work at your own pace, save your progress, and return anytime. If you choose the hybrid approach, we'll connect you with a vetted FDA consultant once your Cruxi project is ready for review.
help Does Cruxi replace my regulatory consultant or RA team?
No. Cruxi is designed to remove grunt work, not judgment. It structures your device information, maps applicable requirements, and drafts sections so that your RA team or consultant can review, correct, and finalize. Human experts remain responsible for strategy, decisions, and sign-off.
gavel Can Cruxi submit directly to FDA or make binding decisions?
No. Cruxi never files submissions, clicks "submit" in eSTAR, or takes regulatory decisions on your behalf. It generates structured, evidence-linked content and recommendations that you can accept, modify, or reject. You stay fully in control of what goes to FDA.
visibility How do I know Cruxi isn't hallucinating or inventing citations?
Cruxi's sophisticated trained multi-chain agents leverage advanced semantic technologies and work over a structured, up-to-date FDA database and your own uploaded documents. These agents use state-of-the-art retrieval and reasoning techniques to ground every claim in verifiable sources. Key statements are linked back to underlying sources (regulations, guidances, public 510(k) summaries, or your files), so you can see where each claim comes from. If the data isn't there, Cruxi doesn't pretend it is—the system is designed to be transparent and traceable, preventing hallucinations through rigorous source verification.
lock Is my confidential device information used to train public AI models?
No. Your device information, documents, and submission drafts are kept within Cruxi's environment and are not used to train public models. They are used solely to serve your projects and to power your workspace.
assignment_turned_in Who is ultimately responsible for the submission?
Always you and your designated regulatory owner (internal RA lead or external consultant). Cruxi exists to significantly enhance quality, minimize human errors and mistakes, make your work faster, more structured, and more transparent. We're not only doing things faster—we're improving accuracy, reducing oversights, and ensuring consistency across all regulatory requirements. However, accountability and final decisions remain with the human experts on the project.
Ready to Start Your 510(k) Submission?
Join medical device companies using a chain of sophisticated trained AI agents to streamline FDA 510(k) submissions. Complete workflow, automated compliance, expert guidance—all in one platform.
Start Your 510(k) Submission